









![]()
GaussView
| What is GaussView? |
GaussView is an affordable, full-featured graphical user interface for Gaussian 98. With GaussView you can construct molecular systems of interest quickly and efficiently using its molecule building facility. You can also set up and run Gaussian calculations right from the interface, and monitor their progress as they run. When a calculation has completed, you can use GaussView to examine a variety of results graphically via its advanced visualization facilities.
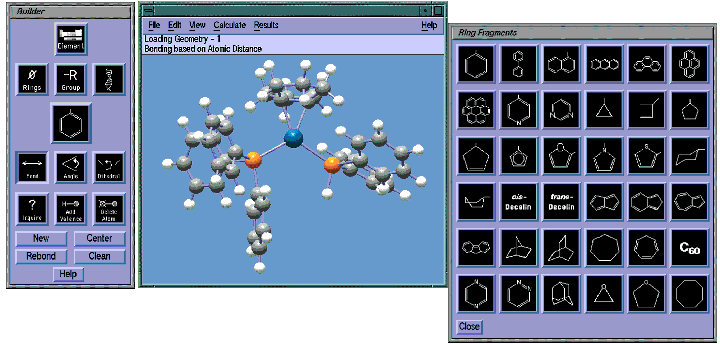
GaussView's Builder palette makes constructing molecules simple and fast. The molecule in the illustration is a platinum-olefin complex. This class of compounds are interesting in that the PtR2group has the ability to stabilize strained olefins upon formation of the complex. Here, we are in the process of completing the molecule by adding a benzene ring to the phosphorous atom on the right. We do so by selecting the desired ring from GaussView's Rings palette and then clicking on the existing hydrogen atom. We can then adjust the angle of the ring using other facilities on the Builder palette.
| Powerful Molecule Building and Viewing Capabilities |
GaussView incorporates an excellent molecule builder which enables even very large molecules to be rapidly sketched in and then examined in three dimensions. It includes the following features:
- Build molecules by atom, ring, group and amino acid. All amino acids are
available in their neutral, N-terminated and C-terminated forms.
- Import molecules from other sources by simply opening them with
GaussView. You can also optionally add hydrogens automatically to
structures originating from PDB files with excellent reliability.
- Examine or modify any structure parameter by clicking on the associated
atoms and using an intuitive dialog box.
- Rotate even the largest molecules in three dimensions. Molecular rotation,
translation and zooming are all accomplished via simple mouse operations.
- View molecules in a variety of customizable display modes: wireframe, tubes and ball and stick.
| Setting up Gaussian Jobs |
GaussView's Gaussian Calculation Setup window allows you to specify any type of Gaussian calculation in a simple, straightforward manner. All of the features of Gaussian 98 are supported by the GaussView interface.
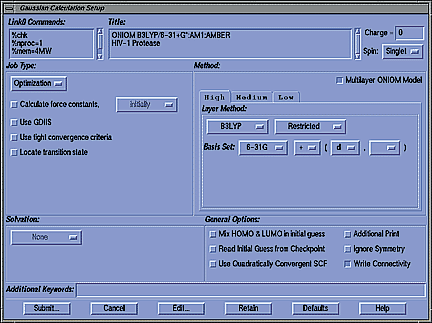
- Appropriate Link 0 Commands are generated automatically by
GaussView, and you can add to or modify them as desired.
- When the desired job type is selected from this pop-up menu, the
appropriate options appear automatically in the area below it.
- The Solvation area allows you to select and specify parameters for any of
Gaussian's available SCRF models.
- For advanced users, the Additional Keywords area allows you to place any
Gaussian keyword into the route section of your job. Once the form is
filled out, you can use the Edit button to open the generated input file in a
text editor for further customization, and the Submit button to begin execution
of the job.
- GaussView fills in the charge and spin multiplicity for you.
- The Method area allows you to specify the desired model chemistry-basis set
and theoretical method-for the job. In this example, we are setting up an
three-layer ONIOM calculation (selected via the corresponding check
box). Here, we specify the model chemistry for the high accuracy layer.
- The General Options area gives you quick access to several commonly-used general purpose options via a single mouse click.
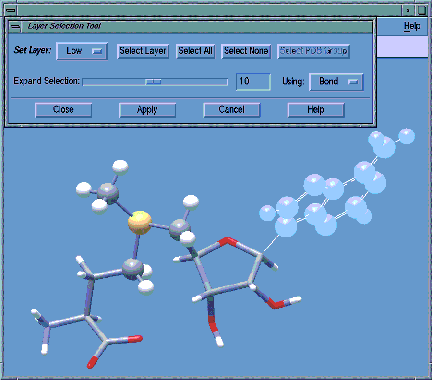
| Features for Setting Up ONIOM Jobs |
- GaussView makes defining the various layers of an
ONIOM calculation straightforward. Atoms can be assigned to layers via
the normal graphical molecule builder interface, and layers defined in existing
Gaussian command files are recognized and preserved.
- Atoms can be selected for layer inclusion in several ways, including by
bond attachment proximity to a specified atom, absolute distance from a
specified atom and placement within a defined group within the originating PDB
file.
- In this example, we are in the process of assigning the selected ring and attached groups to the low accuracy layer via the Layer Selection dialog. GaussView displays the atoms assigned to each layer using a different molecule display mode, in this case, ball and stick for the high layer and tubes for the medium layer.
| Visualizing Gaussian Results |
Gauss View can graphically display a variety of Gaussian calculation results, including the following:
- Optimized molecular structures
- Molecular orbitals
- Atomic charges
- Electron density surfaces from any computed density
- Electrostatic potential surfaces
- NMR shielding density
- Animation of the normal modes corresponding to vibrational frequencies
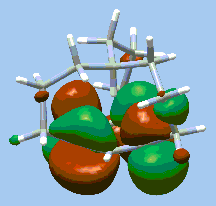 |
Highest occupied molecular orbital for in-[3(4,10)] [7] metacyclophane. The synthesis and characterization of this molecule are described in "Synthesis of in-[3(4,10)] [7] Metacyclophane: Projection of an Aliphatic Hydrogen Toward the Center of an Aromatic Ring." R. A. Pascal, Jr., R.B. Grossman, and D. Van Engen, J. Am. Chem. Soc. 109, 6878 (1987). |
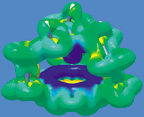
Surfaces may be displayed in solid, translucent and wire-mesh modes. Surfaces can also be colored by a separate property. For example the illustrations below show the electrostatic potential-painted charge density surface for chloroform in both the solid (left) and translucent (right) display modes:
| Presentation and Publication-Quality Output |
GaussView can produce graphical output containing any of the images that it generates. PostScript, JPEG and TIFF formats are supported. You can send such images to a printer for an immediate hard copy or export them to an external file for subsequent inclusion in documents or modification with graphical editing software.
| Studying Magnetic Properties |
Here we present a small study using Gaussian and GaussView illustrating how the two may be used in combination to investigate molecular systems.
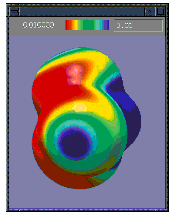
The NMR shielding density for the methine proton of in-[3(4,10)] [7] metacyclophane is shown below (surface on the right), plotted on an isosurface of current density magnitude. (The molecule itself is pictured at the left.) Shielding density increases from red (deshielding) to blue (shielding). It was computed using Gaussian's NMR facility and visualized in GaussView.
The current density, which determines NMR shieldings by the Biot-Savart law, is induced by an external magnetic field parallel to the C3 axis and leads to an unusually large shielding of the methine proton. In accord with the classic ring current model, this result is primarily due to the strong diamagnetic phenyl ring currents and the location of the methine proton, which is calculated to be only 1.70 Angstroms above the phenyl ring. The calculated shielding anisotropy for the methine proton is 23.9 ppm while the calculated isotropic chemical shift is -4.4 ppm relative to TMS, in good agreement with the experimental value of -4.0 ppm.
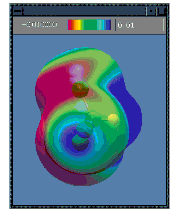
We can compare these shielding densities to those for a phenyl proton in the same compound, shown in the illustration on the left. As before, the shielding density increases from red (deshielding) to blue (shielding). The current density is induced by an external magnetic filed parallel to the C3 axis and leads to a -3 ppm deshielding contribution to the phenyl proton from the bonded carbon and the two neighboring carbons.
The phenyl proton shielding is in sharp contrast to that for the methine proton (left the phenyl ring, along the C3 axis), where the shielding contribution from its own atom is the same as that for the phenyl proton. However, the bonded carbon shields the methine proton by over 5 ppm, and each phenyl ring carbon shields the methine proton by over 3 ppm, leading to a very large shielding of the methine proton. Once again, in accord with the classic ring current model, these results are primarily due to the strong diamagnetic phenyl ring currents and the location of the phenyl and methine protons.

Computed results and images for the NMR study were generated by James R. Cheeseman (Lorentzian, Inc.) and Roy Dennington and Todd Keith (Semichem, Inc.).
Answers to Common Questions About
GaussView
GaussView Pricing